Plain English
The creation of new materials is both difficult and expensive. It is very difficult to “see” the structure of these materials over the length scales that they work and very expensive to create prototype materials to test. Whilst experimental physics can use x-rays, high energy electrons or neutrons to infer the structure of these materials, this inference is made much more robust when combined with theoretical predictions of the kinds of structures that can be formed. In a computer we use quantum mechanical calculations to simulate the results of these kinds of experiments, helping to understand materials and suggest new materials with the kind of properties desired.Battery Materials Modelling
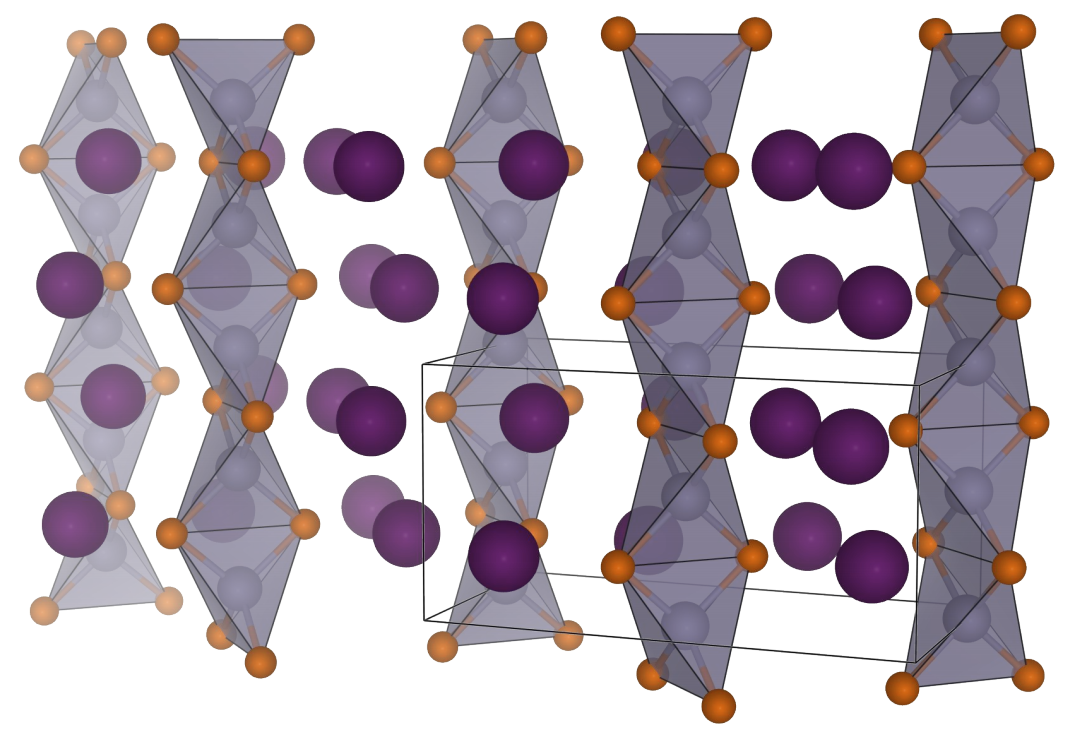
Silicon is the next wonder material for lithium-ion batteries, having a theoretical capacity some 10 times greater than the current graphitic anode. However, there are many obstacles to be overcome such as anode pulverisation, irreversible lithium losses and huge volume changes before the their potential.
Over the course of the project we have found new phases of lithium silicides, lithium germanides, lithium phosphides and lithium sulphides, including new defect and high-pressure phases.
Current Project Members:
-
Matthew Evans
Can Kocer
Angela Harper
Collaborators:
We have a long running collaboration with Prof Clare Grey and Prof Chris Pickard using the ab initio random structure searching method (AIRSS) to predict the stable phases of electrodes that occur as a lithium-ion battery charges.
Encapsulation in Nanotubes
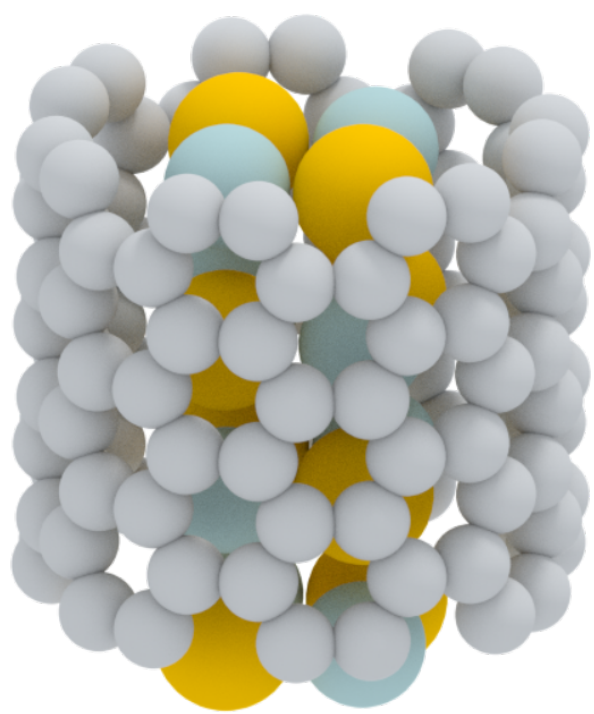
Particularly exciting recent work by Jeremy Sloan has demonstrated the possibility of nanowires undergoing transitions between nano-crystalline structures with markedly different properties, in response to bending strain in the CNT. These “phase change” properties open the way for nanoscale electromechanical switches and non-volatile memory, as well as providing a playground for fundamental studies of phase changes at the smallest length scale possible in a material.
Our aim with the current project, inspired by these results, is to develop a computational modelling capability to aid in interpretation of experiments, understand the origin of the phase change behaviour, and guide our experimental colleagues toward compounds with potentially advantageous properties.
Collaborators
In collaboration with Drs David Quigely and Jeremy Sloan at the University of Warwick. We predict the structure of tiny crystals that can form inside carbon nanotubes.
Structure Prediction
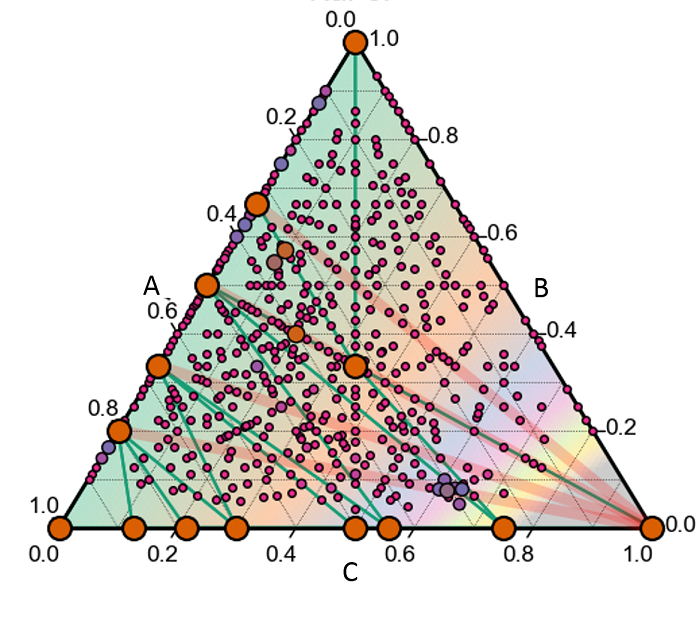
In addition to using structure prediction tools to characterise systems of experimentally relevant materials, such as batteries and nanowires, we also develop our own software for crystal structure prediction which builds off of ab nitio random structure searching (AIRSS). Developing smarter tools for structure prediction is a key component of identifying new materials, and so our group aims to not only be users but developers as well.
Current Project Members
-
Matthew Evans
James Darby
Angela Harper
Software
Theoretical Spectroscopy
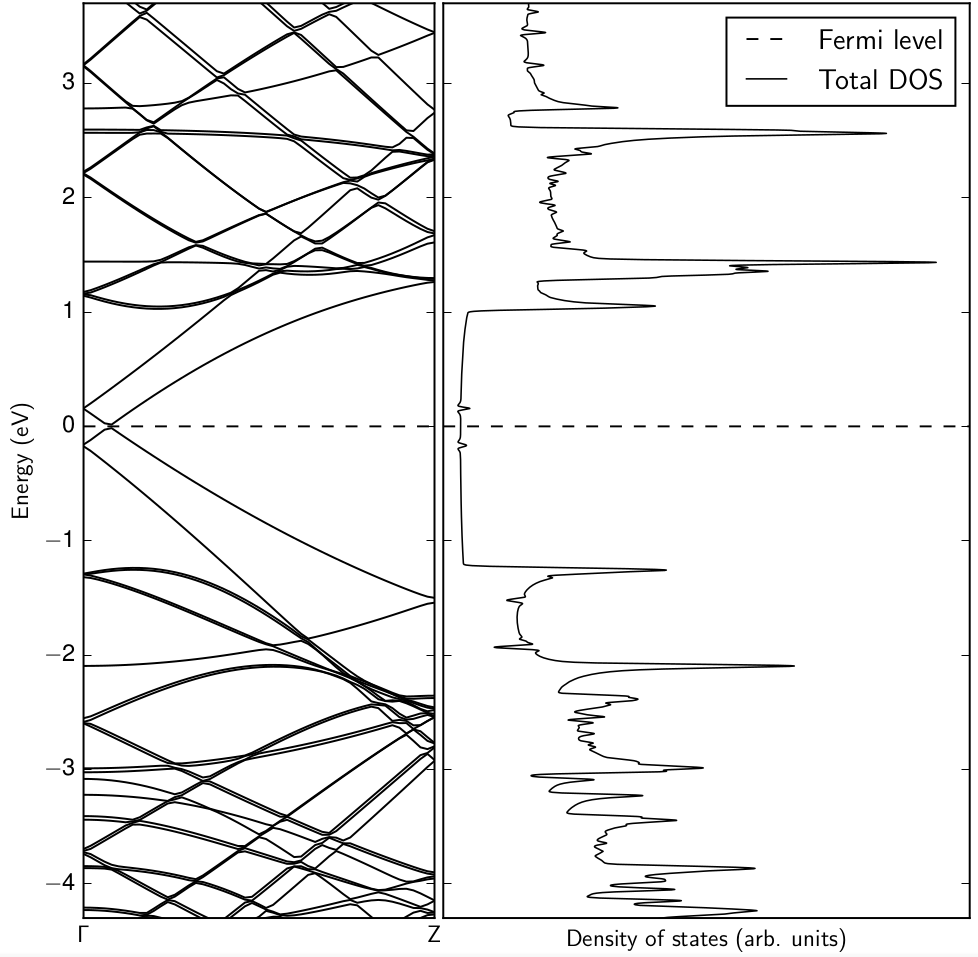
Producing high-quality density of states plots from Kohn-Sham eigenvalues is important for understanding the electronic structure and position of impurity states. Simple Gaussian smearing is ineffective due to the nature of dispersive bulk and localized defect states whereas the linear tetrahedron method is interpolative and will always avoid band crossings.
OptaDOS is a code for calculating optical, core-level excitation spectra along with full, partial and joint electronic density of states (DOS). At present OptaDOSinterfaces with CASTEP and ONETEP output files, although it is extendible to perform calculations on any set of band eigenvalues and their derivatives generated by any electronic structure code.
OptaDOS is open source an can be freely downloaded by the scientific community and is used by many research groups around the world
Current Project Members
-
Andrew Morris
Can Kocer
Collaborators
In collaboration with Prof Jonathan Yates and Dr Rebecca Nicholls at the University of Oxford we author the OptaDOS code.
Software