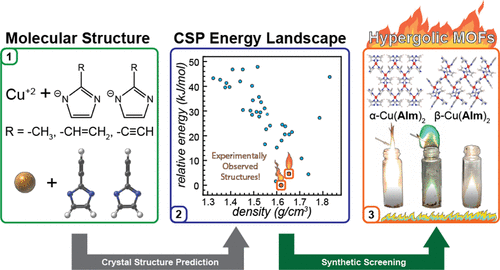
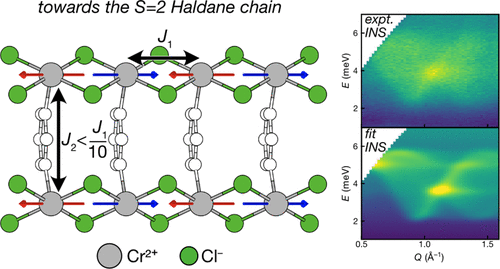
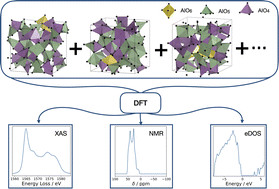
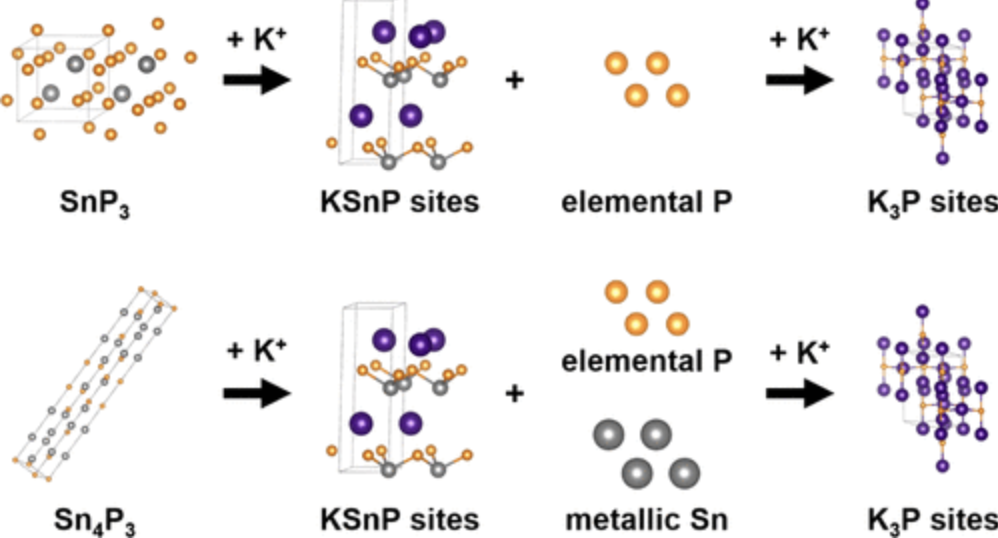
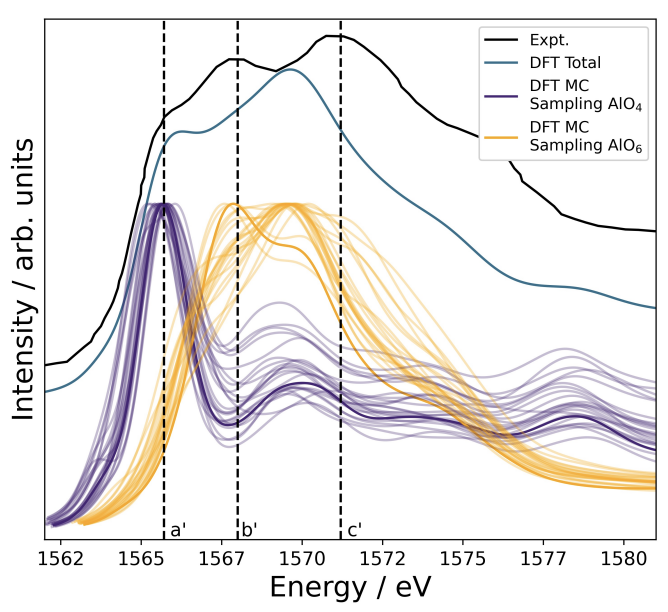
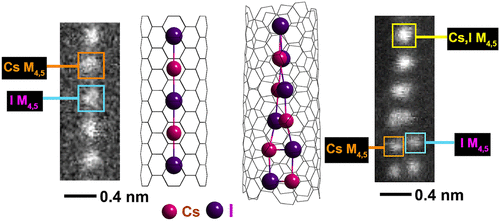
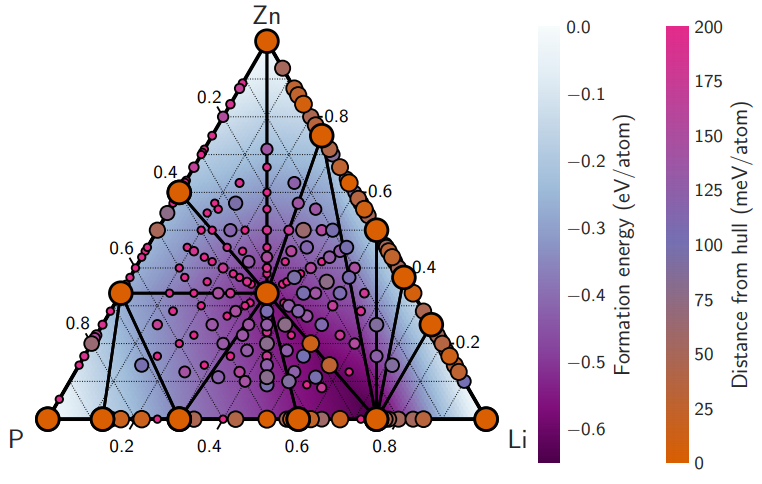
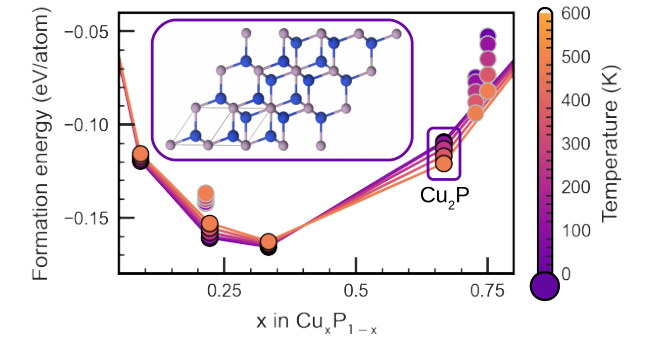
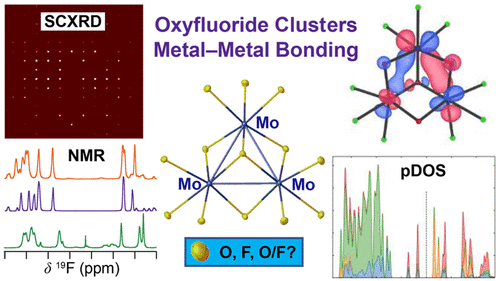
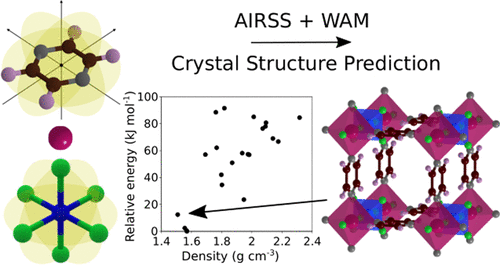
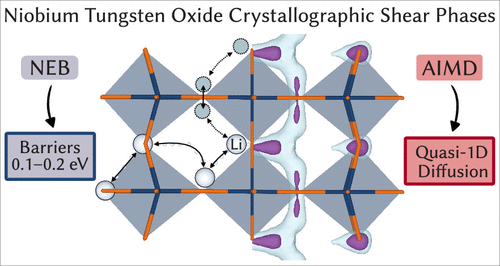
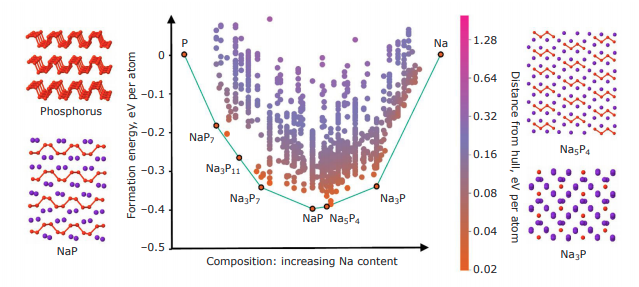
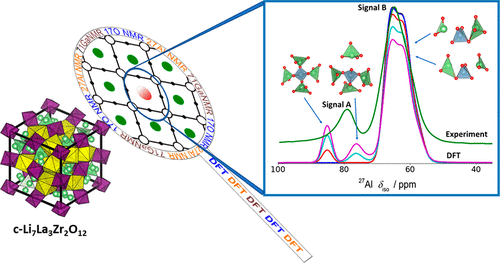
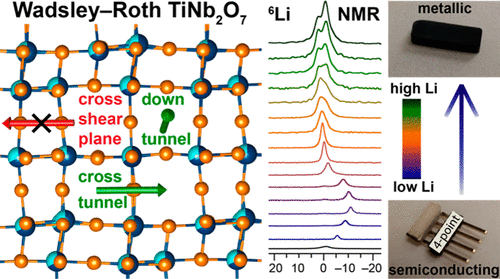
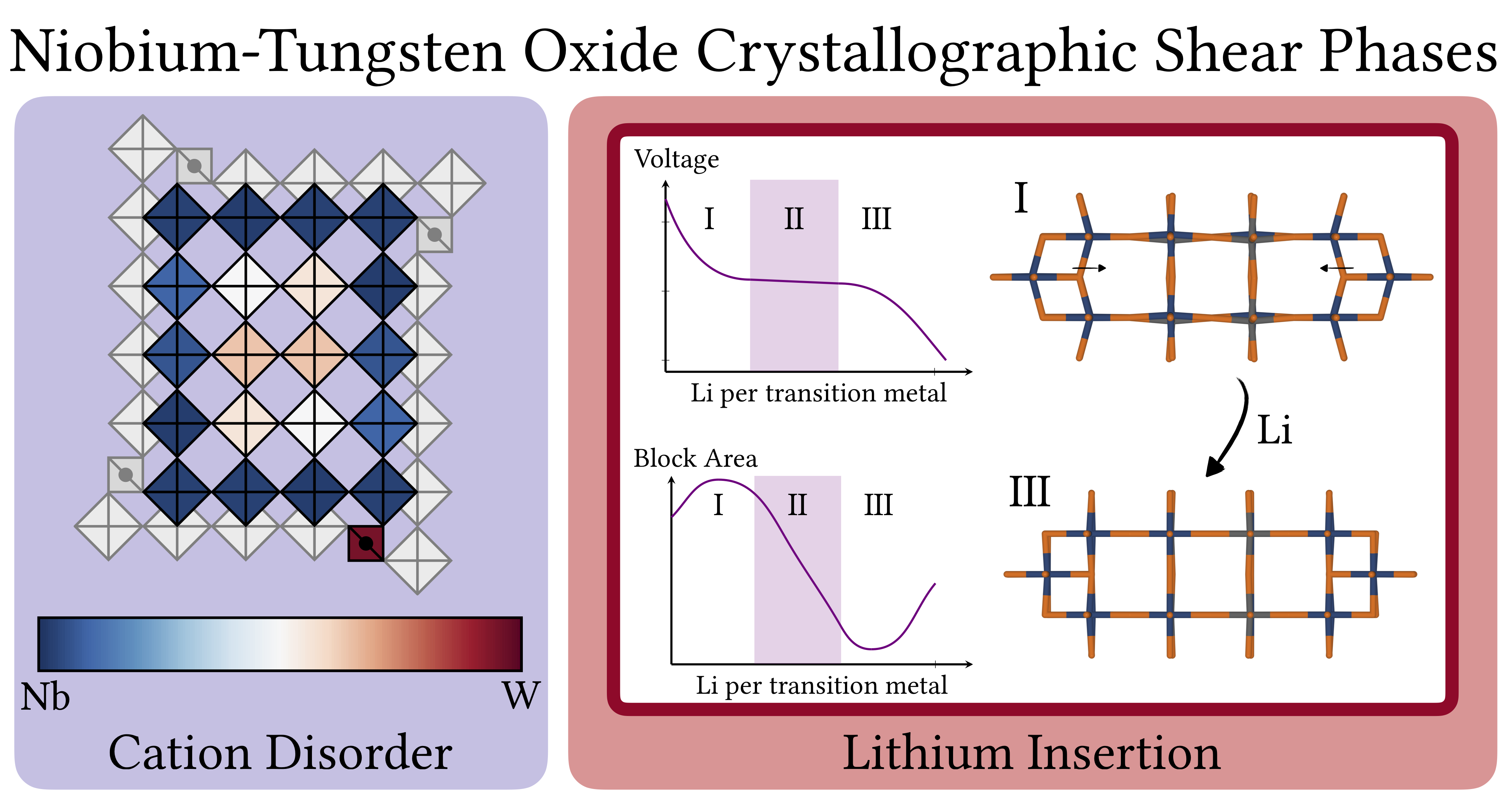
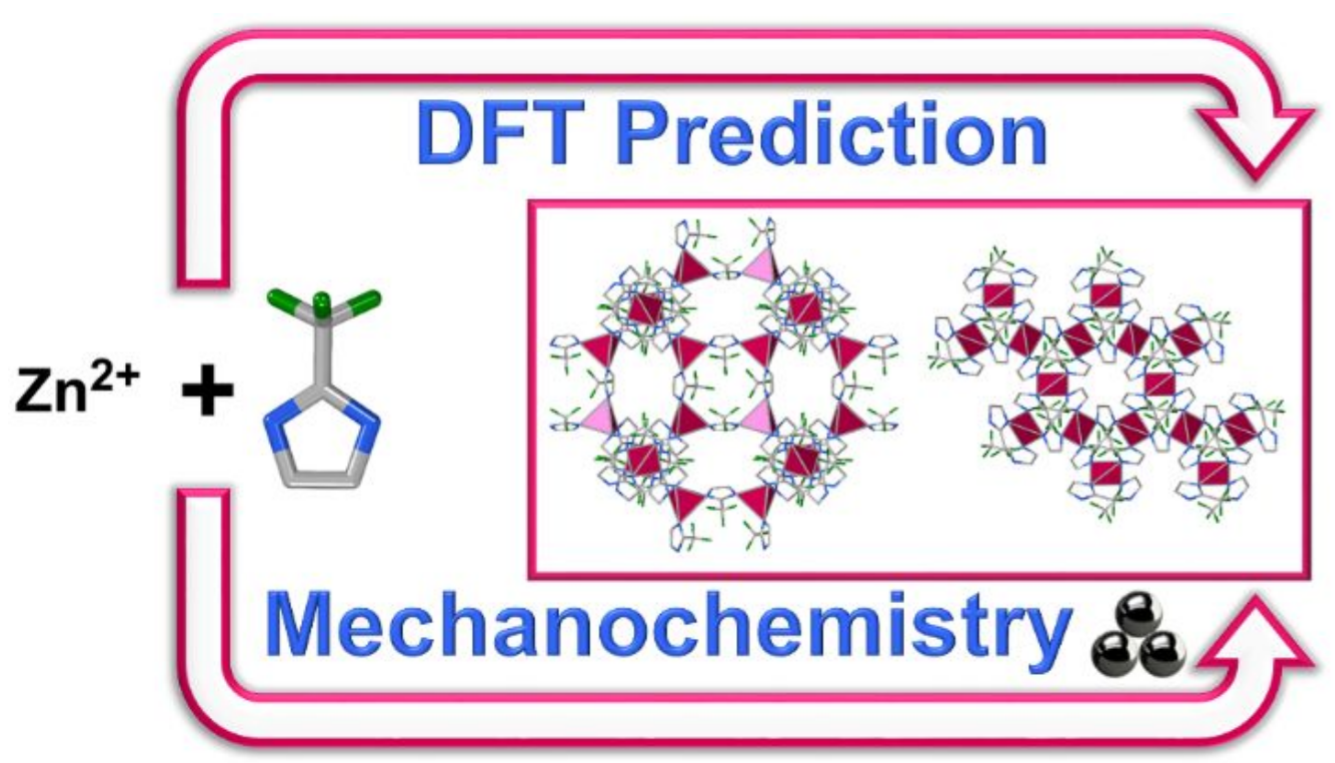
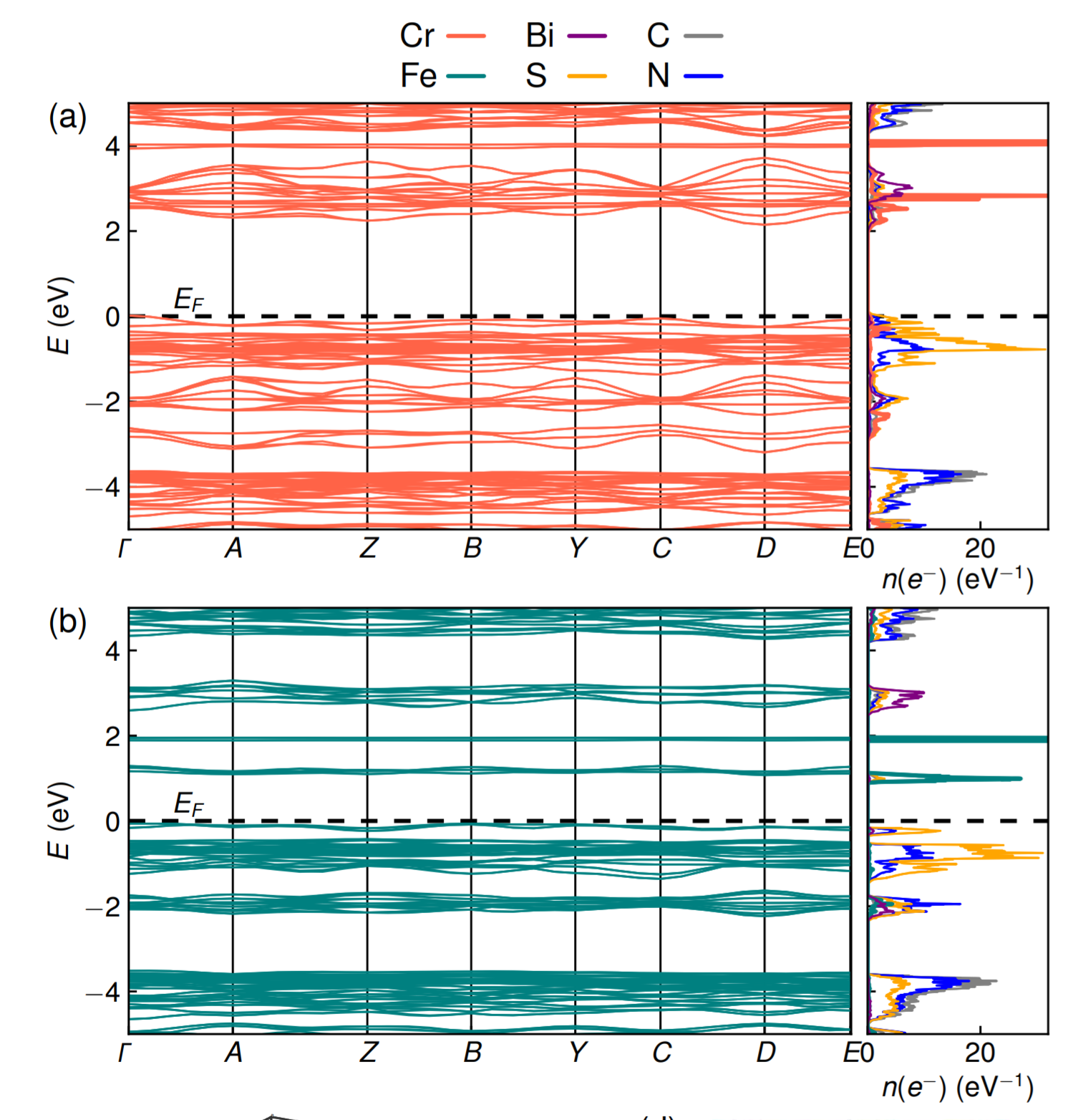
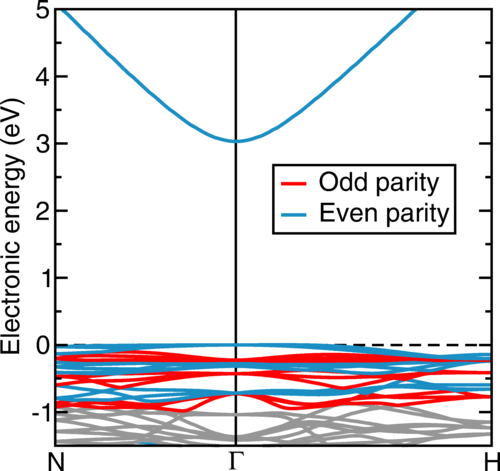
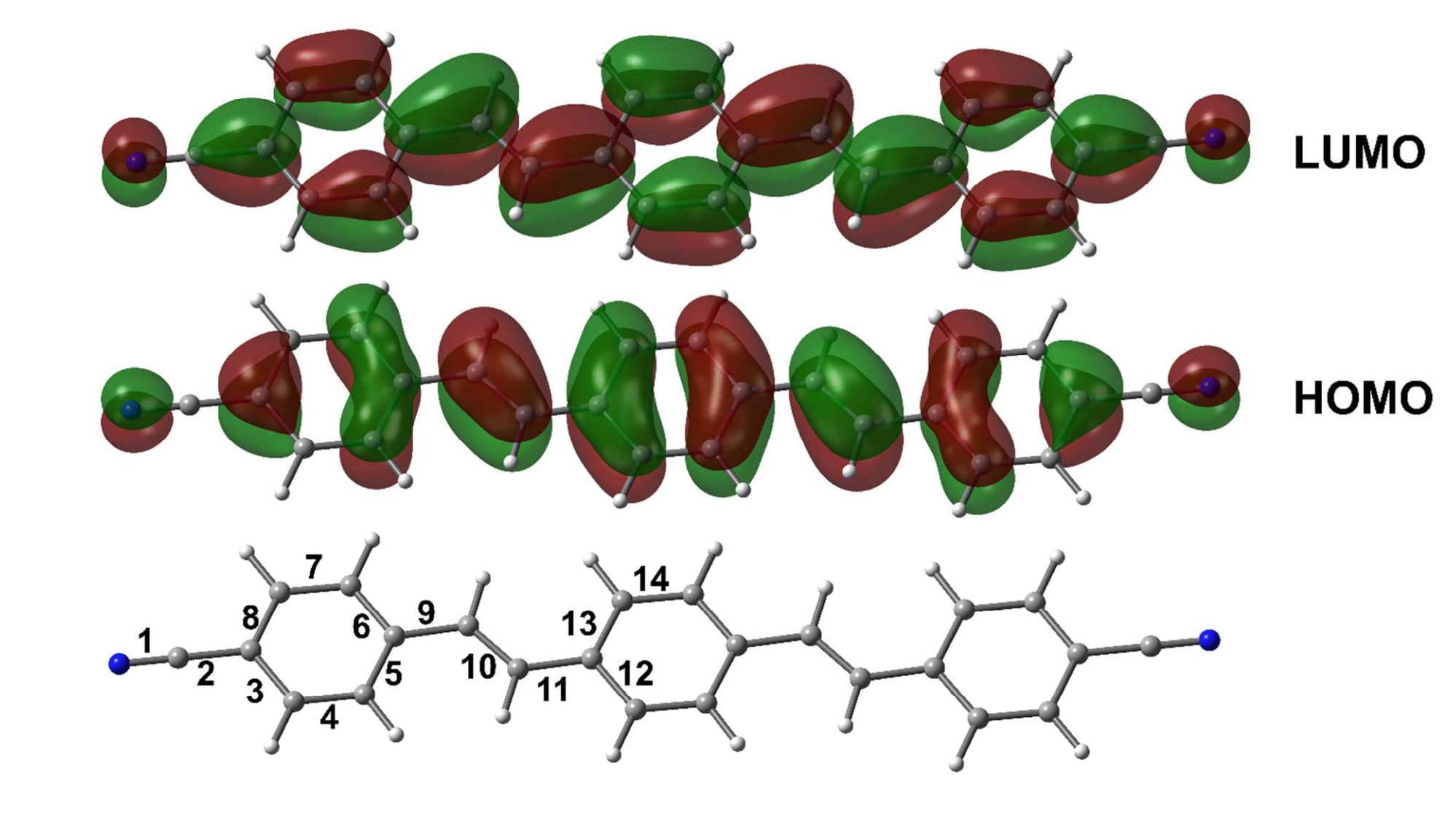
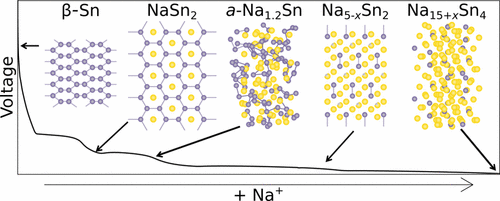
Atoms stick together to make the world around us through a set of different atom-atom interactions called bonds. Without atomic bonding, the universe would be just be one large gas with nothing solid in it at all.
The text-book four types of atomic bonding are ionic (think table salt), covalent (silicon chips), metallic (steels, iron etc) and van der Waals (how geckos stick to the ceiling). However, there is a fifth kind of bonding, called a non-covalent directional interaction. It is responsible for water’s unusual properties such as expanding on freezing (most liquids shrink) and having a high boiling point compared to other molecular liquids.
We care about this fifth kind of bonding as it can stick molecules together into molecular crystals which have a wide variety of uses, for example, capturing carbon dioxide from the atmosphere, detecting trace impurities and toxins, sensing biomolecules and a vast collection of pharmaceuticals. It’s similar to molecular Lego – the more different ways we can stick bricks together, the more useful (and fun!) materials we can make.
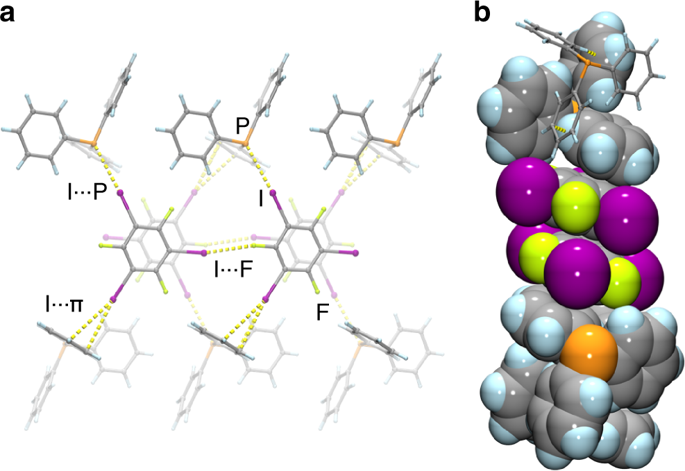
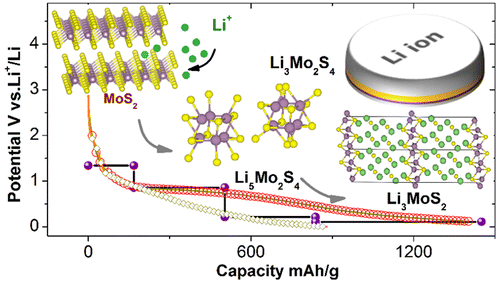
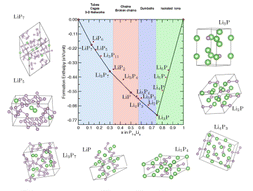
The halogen elements such as fluorine, chlorine, bromine, and iodine can form these fifth-kind bonds. Up until now they were thought to only bond to small, compact atoms like nitrogen or oxygen, limiting the ways molecular crystals could be assembled. In this international collaboration between UK, Canada and Croatia we show that we can make molecular crystals by bonding the halogen iodine to the much heavier phosphorus, arsenic and antimony atoms, some 2, 5 and 8 times more massive than oxygen respectively.
This research paves the way for many new molecular crystals with fantastic new properties. We show, for example, colossal expansion in a crystals with I…Sb bonds on heating.
Full Article